All Research
Baycrest research advances understanding of aging and brain health — from discovery to real-world application — shaping care, education and health systems.

Adjunct Scientists
Our adjunct scientists conduct a wide range of research on aging, brain health and dementia care.
Learn More
Baycrest Academy for Research and Education
The Baycrest Academy for Research and Education advances aging, dementia and brain-health research while training the next generation of professionals.
Learn More
Baycrest Open Science and Open Scholarship
At Baycrest, we believe scientific research and scholarship are strongest when it is open, transparent and accessible.
Learn More
Participate in Research
Learn how to participate in Baycrest studies that look at the brain and aging.
Learn More
Research Ethics Board
The REB reviews all research at Baycrest to ensure it is safe, ethical and protects the rights and well-being of participants.
Learn More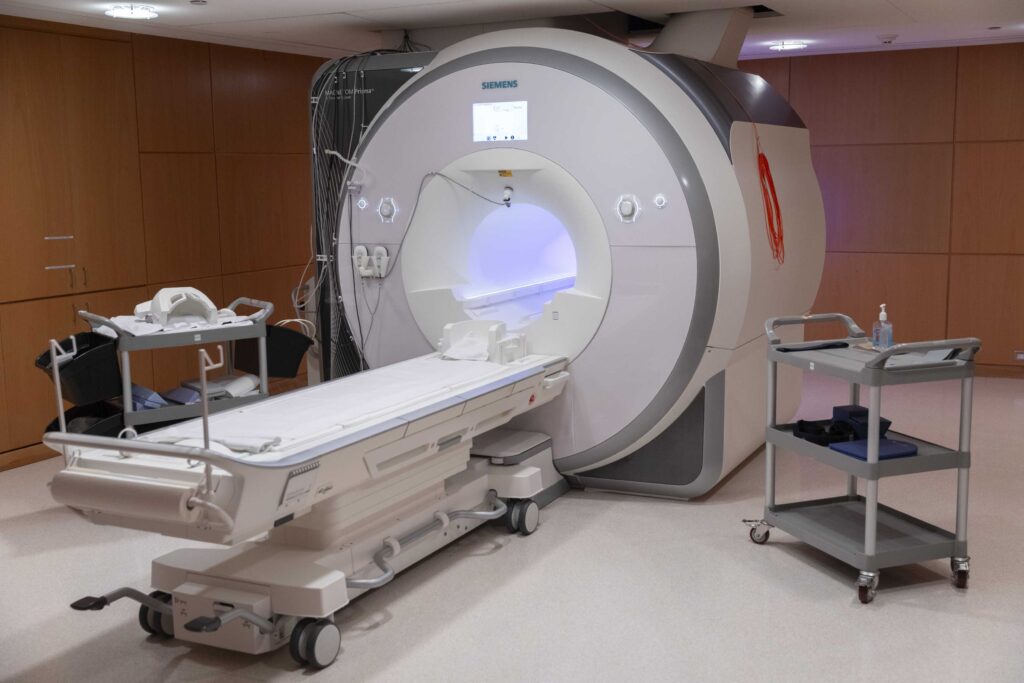
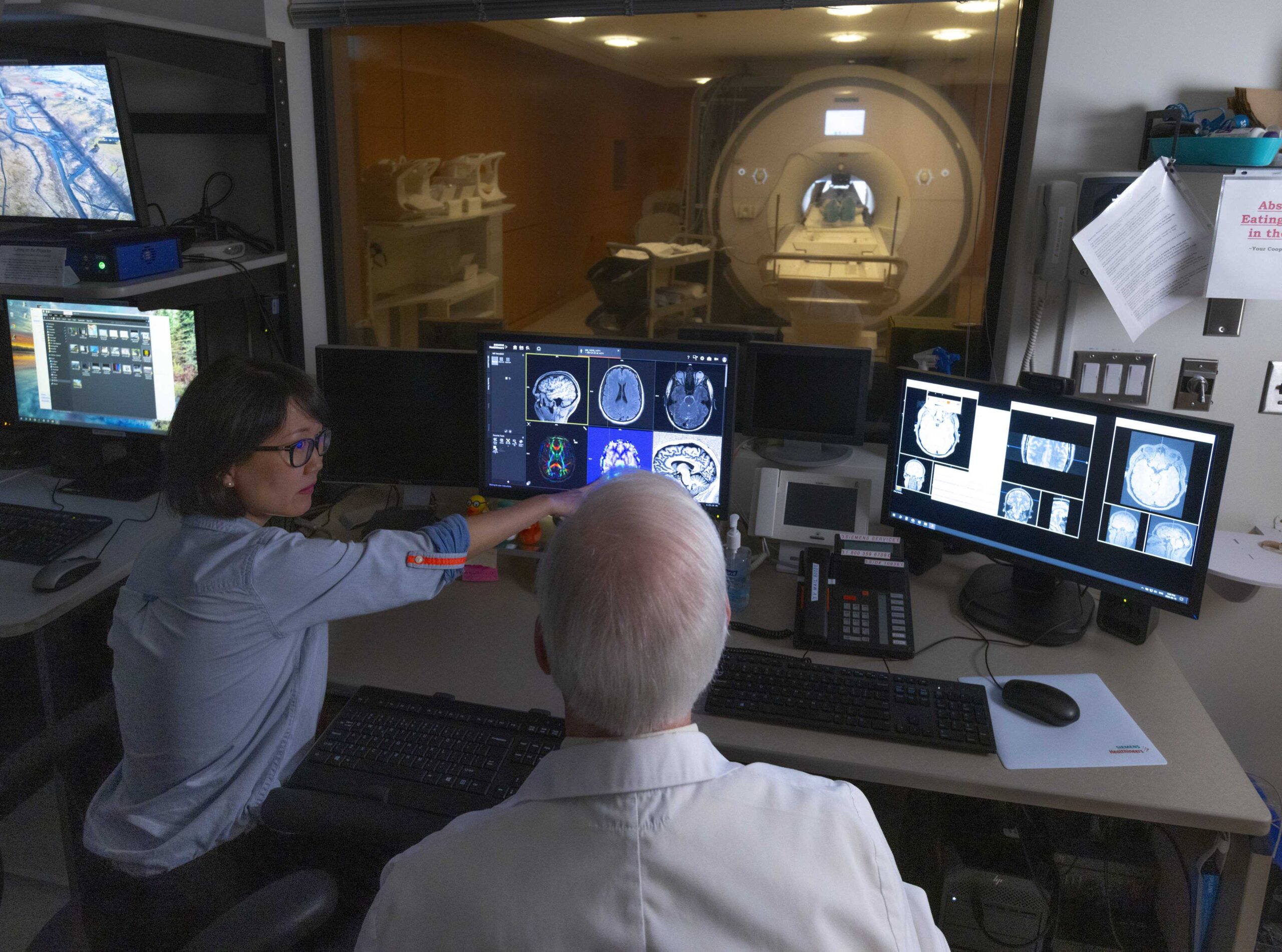
Research Facilities
Learn about the advanced neuroimaging and research technology available at Baycrest to advance research in aging and brain health.
Learn More
Research Themes
Our research focuses on aging and brain health that aims to improve care and quality of life for older adults.
Learn More
Research Training Centre
Baycrest’s Rotman Research Institute offers students and other trainees a wide range of learning opportunities.
Learn More
Research Units
Baycrest research labs advance foundational science in aging and brain health, where discovery informs education, care and real-world impact.
Learn More
Rotman Research Institute
Baycrest’s Rotman Research Institute conducts leading-edge research on aging, brain health and dementia care.
Learn More
Scientists, Scientific Associates and Clinician Associates
Our scientists conduct a wide range of research on aging, brain health and dementia care.
Learn MoreStudents and Learners
Learn about our student placement opportunities with experts in aging and brain health.
Learn More